
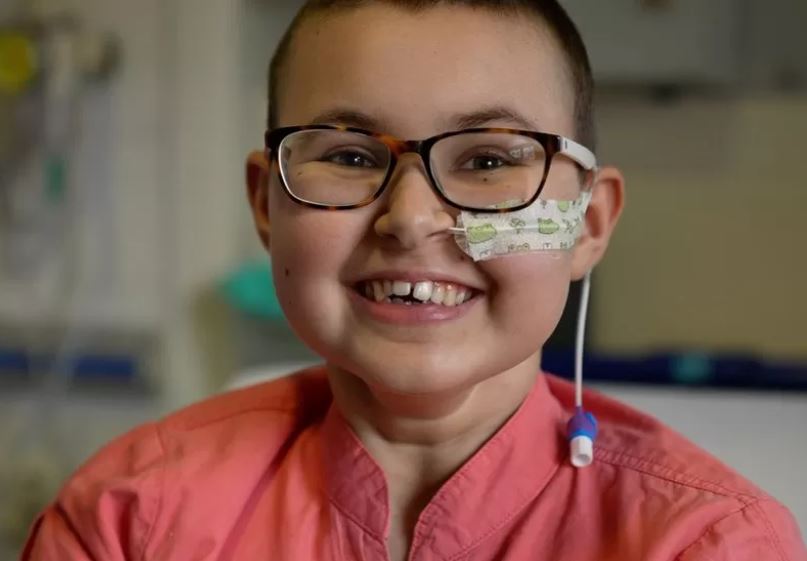
Hace unos días os hablaba en este blog de Daniela, la recién nacida a quien la investigación científica y la medicina habían salvado la vida. Hoy os quiero hablar de otra historia de éxito de la medicina moderna que creo merece conocerse y contarse. Y es una historia que acabamos de descubrir, a raíz de un comunicado de prensa publicado por la University College London (UCL). Alyssa es el nombre de una niña británica de 13 años, diagnosticada con un tipo de leucemia muy agresiva (leucemia linfobástica aguda de células T, conocida por sus siglas en inglés, T-ALL) desde mayo de 2021 que había recibido tratamientos convencionales, sin éxito, y estaba médicamente desahuciada.
En mayo de 2022, un año después de ser diagnosticada, fue ingresada en la unidad de transplantes de médula ósea del hospital pediátrico Great Ormond Street de Londres para ser tratada con un nuevo tipo de terapia CAR-T, una aproximación terapéutica que puede incluir el uso de las herramientas CRISPR de edición genética, pero, en este caso, no se trataba de las herramientas CRISPR tradicionales, sino de una de las variantes aparecidas posteriormente, gracias a la investigación científica básica: los editores de bases. Y con un uso innovador. La utilización de editores de bases concebidos no para «corregir» genes sino para todo lo contrario, para inactivarlos.

La inactivación que puede obtenerse con los editores de bases es mucho más segura que la que podría obtenerse con las herramientas CRISPR/Cas9 o CRISPR 1.0 y sus conocidos problemas de inactivación de genes parecidos (off target) y la diversidad de variantes genéticas que pueden generarse (on target o mosaicismo), que prácticamente desaparecen o se reducen muchísimo con los editores de bases (o con el prime editing / la edición de calidad). Pero el prime editing todavía no ha llegado a la clínica, mientras que el base editing, los editores de bases, sí. Y lo que llega a los hospitales no son las aplicaciones que uno pensaría como obvias (substituir la letra mutada por la correcta para reactivar el gen afuncional) sino el uso de editores de bases para mutar, para inactivar específicamente determinados genes. Y esto puede hacerse con editores de bases porque estos funcionan sin cortar las dos cadenas del ADN, y pueden propiciar la aparición de codones de terminación, codones stop. Esto es un salto conceptual importante que puede tener trascendencia en otras muchas enfermedades y va a revolucionar cómo la evolución de las herramientas originales CRISPR/Cas9, transformadas en Editores de Bases, van a llegar a la clínica.
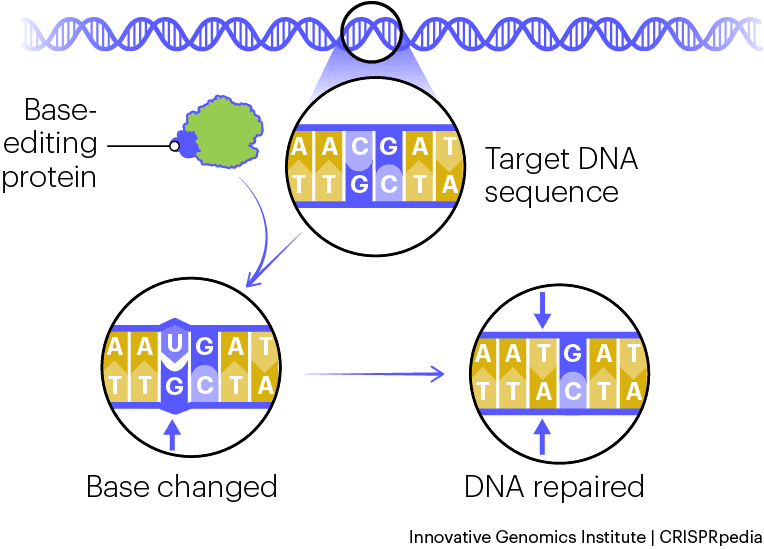
El uso de los editores de bases para inactivar un gen induciendo la aparición de un codón de parada (Stop) ya fue explorado hace algo más de un año por un equipo de investigadores que demostró que podían usarse editores de bases para inactivar el gen PCSK9, y con ello conseguir una reducción del colesterol circulante en la sangre. El principio es muy sencillo de entender. Los genes portan la información necesaria para fabricar proteínas. El ADN, de doble cadena, se transcribe (se convierte en) a ARN, de una cadena, en el que las T se substituyen por las U. Y, el ARN sale del núcleo y es leído por los ribosomas para fabricar las proteínas, lo que conocemos como traducción. El código genético que usan los ribosomas esta basado en grupos de tres letras, trinucleótidos, también llamados codones, y cada uno de los 64 posibles codones que podemos obtener combinando las cuatro bases A, G, C y U (4x4x4=64) codifica un aminoácido distinto o algunas señales de inicio o parada de la traducción. Por ejemplo, el codón UAU codifica el aminoácido tirosina (Tyr) pero si editamos la última U y la convertimos en una A (UAU–>UAA) entonces el codón UAA es un codón de parada, de Stop. Y se detiene la traducción de la proteína, obteniéndose una proteína truncada que habitualmente no será funcional. Para que esto ocurra tal y como lo explico deben usarse los editores de bases que se encargarán de convertir la T inicial del ADN (que aparece como U en el ARN) en una A. Esta es la aproximación que estos investigadores británicos han usado para tratar a Alyssa con editores de bases: usarlos para generar codones de parada en medio de los genes cuya función necesitaban inactivar.
Alyssa recibió un transplante alogénico (no con sus células, sino con las células de otra persona) de linfocitos T de un donante sano (proporcionadas por el registro Anthony Nolan, una iniciativa ciudadana que promueve las donaciones de médula ósea fundada por Shirley Nolan cuando no pudo encontrar células compatibles con las de su hijo Anthony, que da nombre a esta fundación) que habían sido editadas, modificadas genéticamente, en el laboratorio.
Esas células T recibieron cuatro modificaciones, necesarias antes de ser infundidas a Alyssa:
- Inactivación del gen del receptor de células T (TCR) para hacerlas universales (y para poder añadir luego un receptor específico, CAR).
- Inactivación del gen CD7 para evitar que la terapia anti-células T acabe también con ellas mismas.
- Inactivación del gen CD52, que es la diana de potentes medicamentos antitumorales que se administran a estos pacientes, para que no fueran afectadas por estos tratamientos.
- Adición de un CAR (chimeric antigen receptor, receptor quimérico de antígeno) que reconoce a la proteína CD7 presente en la superficie de las células T cancerosas, causantes de la leucemia T-ALL de Alyssa.
Los tres primeros pasos, que teóricamente podrían haberse abordado con herramientas CRISPR tradicionales (con el consabido riesgo de editar otros genes similares y/o de generar una diversidad de alelos innecesarios) son los que se han realizado mediante los editores de bases. Se han usado editores de bases programados para inactivar esos tres genes (TCR, CD7 y CD52) introduciendo cambios en una base y creando codones de stop prematuros, una forma muy específica, y limpia, de inactivar genes con un menor riesgo de efectos colaterales. El cuarto y último paso fue la reintroducción de un nuevo receptor de antígeno quimérico, CAR, específico para reconocer la proteína CD7 en la superficie de las células T tumorales y destruirlas. Todo el proceso está magníficamente explicado gráficamente en este artículo de la BBC.
Al administrar estas células T modificadas con editores de bases a la paciente consiguieron eliminar completamente las células T tumorales. Seguidamente, un més después, la paciente recibió un segundo trasplante de médula ósea que restauró su sistema inmunitario, con células sanas, no modificadas genéticamente. Seis meses más tarde la niña ya está curada, en su casa, siguiendo los controles rutinarios que siguen todas las personas trasplantadas.
Los investigadores y médicos involucrados en este tratamiento pionero han presentado los resultados del tratamiento de Alyssa en el congreso anual de la American Society of Haematology en New Orleans, USA. Este es un primer caso, un primer paciente tratado con éxito, de muchos otros que esperemos seguirán a Alyssa. Podéis leer los detalles del ensayo clínico asociado, que sigue reclutando pacientes, y otros comentarios sobre este tratamiento experimental pionero en New Scientist y en The Guardian.
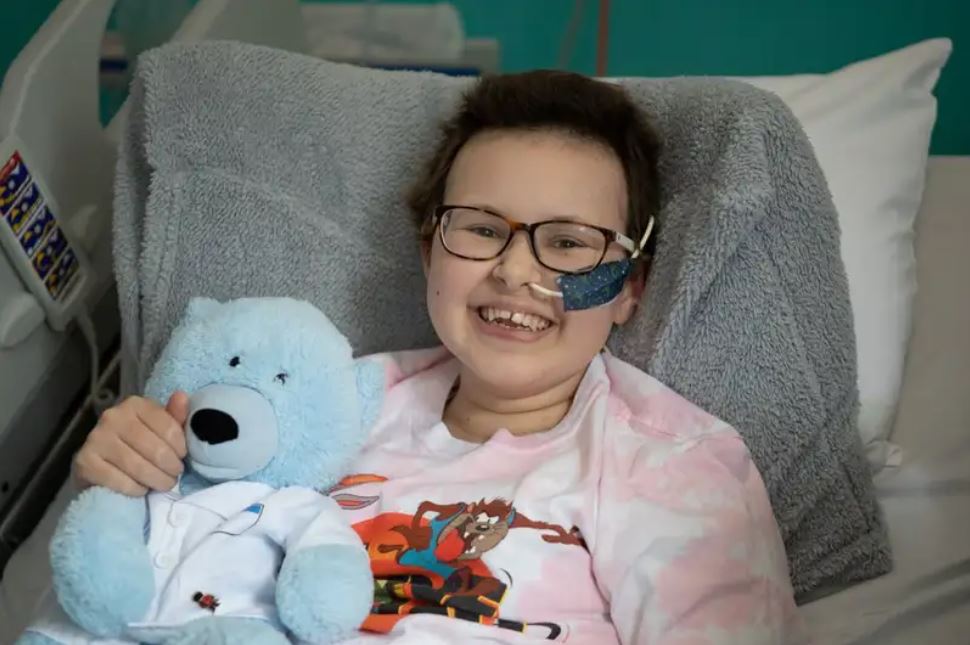
Alyssa es pues la primera paciente que podemos decir que se ha beneficiado, se ha curado, gracias a los editores de bases. La primera persona tratada con éxito con estas variantes de CRISPR que fueron propuestas por la imaginación desbordante de David R. Liu (Broad Institute) y publicadas hace apenas seis (6) años, en 2016. Esperemos que Alyssa sea la primera paciente de muchas otras que deberían poder beneficiarse de este gran éxito de la ciencia básica y de la medicina.
Esta misma semana Fyodor Urnov se preguntaba en una tribuna de opinión que escribía en el New York Times ¿por qué no estábamos ya aplicando las tecnologías CRISPR en la clínica? ¿Por qué no hacíamos todo lo posible por acercar las terapias CRISPR a todo el mundo que las necesitara? En Londres ya han empezado.
Se trata de un procedimiento asumible económicamente? Me explico, no estamos hablando de 2 millones de Euros por paciente.
Ignoro el coste de este procedimiento, que está en fase experimental. Es parte de un ensayo clínico actualmente en marcha. No está, todavía, aprobado como tratamiento.
Es fascinante (al menos para mí que tengo 85 años) ver los avances científicos en estos últimos años. Felicitaciones y que Dios bendiga a los responsables de estos avances.
Impresionante y magnífica noticia expuesta en su artículo Sr. Montoliu. La ciència va avanzando ??
En mi caso, al igual que mi madre y hermana, presentamos una mutación mitocondrial en el gen MT-CYB, en la posición 15674T>C, con cambio nucleotidico Ser310Pro. El fenotipo más evidente es una neuropatía óptica con clínica similar a la neuropatia òptica de Leber (LHON), haciendo que perdamos rápidamente capacidad visual. También hay otros fenotipos de naturaleza neuroesqueletica.
Como se ha comentado, al ser enfermedades raras, el coste de estos tratamientos por desgracia, es inasumible en muchos casos.
Por otro lado, el tratamiento de mutaciones mitocondriales aún no está conseguido o validado, cierto??
El acceso a las mitocondrias es complicado debido a su naturaleza.
Hay una posible previsión o esperanza para el tratamiento genético de mutaciones mitocondriales??
Hay que seguir apoyando este tipo de investigaciones y a quienes las llevan a cabo, pues son el futuro y esperanza de muchas personas afectadas por mutaciones genéticas, tanto nucleares como mitocondriales.
Saludos cordiales
David R. Liu es quien ha llevado los beneficios potenciales de la edición genética mejorada (los editores de bases) a las mitocondrias.
Y sin usar CRISPR, sino TALEN, las herramientas anteriores
Estos dos artículos son buen ejemplo de ello:
https://pubmed.ncbi.nlm.nih.gov/32641830/
https://pubmed.ncbi.nlm.nih.gov/35379961/
???????? Una gran revolución médica en ciernes, cada vez más tangible.