

Con el titular de este artículo empiezan muchos de los mensajes que recibimos regularmente quienes nos dedicamos a investigar sobre enfermedades raras. Madres y padres que escudriñan internet en busca de quien les pueda ayudar a entender lo que le ocurre a su hijo, lo que le pasa a su hija. Algo que no estaba en sus planes cuando decidieron tener descendencia. Algo con lo que no contaban. Pero algo que, en el mejor de los casos, convivirá con ellos durante toda su vida, la de ellos y la de sus hijos afectados por alguna de las llamadas enfermedades raras.
Ante todo, información es lo que piden estos padres. Saber de qué se trata. Conocer por qué su hijo es una de las aproximadamente tres millones de personas que estimamos existen en España conviviendo con alguna enfermedad rara. La Medicina actual tiene constancia de unas 18.000 enfermedades. Un tercio de ellas son raras, poco frecuentes. En su gran mayoría de origen genético, por eso las llamamos congénitas, y, lo que las hace más terribles todavía, generalmente afectan o se manifiestan en niños, desde su nacimiento. El único nexo en común que mantienen es su baja frecuencia en la población, arbitrariamente definida en Europa como toda patología que afecte a menos de una de cada dos mil personas nacidas. Lo cual es un verdadero cajón de sastre, donde tienen cabida alteraciones musculares, óseas, metabólicas, nerviosas, sensoriales, cardiovasculares, digestivas, inmunológicas, de comportamiento… que pueden cursar con discapacidades físicas o psíquicas con una presentación extremadamente variable, desde disfunciones que apenas se perciben hasta trastornos que modifican significativamente la calidad de vida de las personas que las padecen y de sus familiares.
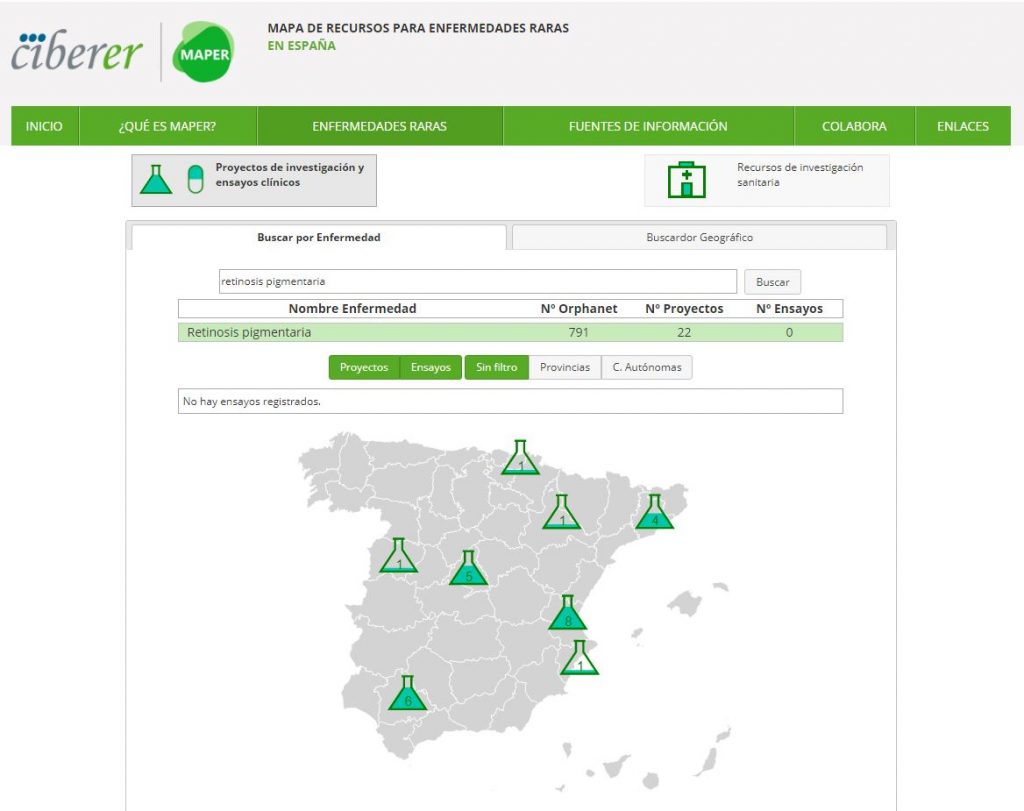
Es imposible ser especialista en enfermedades raras. A lo más que un investigador puede aspirar es a especializarse en alguna de las miles de enfermedades raras, o en algún grupo de enfermedades poco frecuentes que estén relacionadas. Por eso son tan importantes las iniciativas nacionales e internacionales que agrupan a científicos con voluntad de explorar la causa y el remedio para alguna de estas 6.000 enfermedades raras. En España, en el Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER), dependiente del Instituto de Salud Carlos III, convivimos 60 laboratorios y 18 grupos clínicos vinculados que, globalmente, directa o indirectamente estudiamos alrededor de 2.500 de estas enfermedades poco frecuentes. Probablemente, además de los muchos resultados y publicaciones ya obtenidas desde el CIBERER, una de las aplicaciones de la que nos sentimos más orgullosos, aunque sorprendentemente poco conocida, es el MAPER, el mapa de proyectos de investigación y ensayos clínicos en Enfermedades Raras en España, que nos indica dónde y quién trabaja con qué enfermedad rara en nuestro país. Encaminarles hacia él suele ser mi respuesta habitual a los mensajes que recibo de familias con alguno de sus miembros afectados por alguna enfermedad rara. No hay nada mejor para estas familias que ponerlas en contacto con algún grupo de investigación que esté o haya trabajado en esa patología en la que están interesados, que les entienda, les dé información y consuelo y les aporte el estado actual de nuestro conocimiento científico relativo a esa enfermedad. Esa enfermedad dejará de ser rara para esa familia, y se convertirá en un aspecto central de sus vidas, las de todos los miembros de la familia, que girarán alrededor de esa persona con esa enfermedad rara y de sus necesidades.
La gran mayoría de las enfermedades raras están causadas por el funcionamiento anómalo de alguno de nuestros 20.000 genes. Habitualmente tenemos dos copias de cada gen, una heredada de nuestra madre y otra heredada de nuestro padre. La excepción la encontramos en los genes que están en los cromosomas sexuales X e Y, el vigesimotercer par que todos tenemos en nuestras células. En varones, que son XY, el cromosoma X lo heredan entero de la madre, y el Y directamente del padre, mientras que las mujeres, que son XX, heredan un cromosoma X de cada progenitor. Por eso los varones suelen presentar con mayor frecuencia enfermedades asociadas al mal funcionamiento de algún gen situado en el cromosoma X, alteración que no puede compensarse con la otra copia del gen, que no existe en el cromosoma Y. Son las llamadas enfermedades con herencia ligada al sexo como las hemofilias, el daltonismo o el albinismo ocular.
En general, para aquellos genes que no están en los cromosomas sexuales, que son la mayoría, mientras heredemos alguna de las dos copias, o las dos, que funcionen correctamente vamos bien. Pero si coinciden en una persona dos copias anómalas del mismo gen no hay compensación posible, y entonces se establece la enfermedad. Y siempre llega por sorpresa. El azar une a una pareja que habitualmente ignoran que son portadores, los dos, de mutaciones en el mismo gen. Ninguno de los dos miembros de la pareja manifiesta la enfermedad porque tienen una copia correcta que permite mantener la función génica. Pero, en cada embarazo, la ruleta genética gira. Cada progenitor tiene una probabilidad del 50% de transmitir a su hijo la copia anómala del gen. Por eso, (0,5 x 0,5 = 0, 25) con un 25% de probabilidad, en cada embarazo, existe la posibilidad que nazca un hijo sin esa función génica, un niño que pasará a engrosar las estadísticas de personas con enfermedades raras.
¿Qué podemos hacer cuando se presenta una enfermedad rara? ¿En qué investigamos quienes nos dedicamos profesionalmente al estudio de estas patologías? Fundamentalmente en dos aspectos: diagnóstico y terapia. Primero de todo saber qué gen o genes están afectados y cuáles son las mutaciones causantes de la enfermedad. Seguidamente, una vez descubierta la función génica que falta, intentar desarrollar algún tratamiento, alguna terapia, que alivie los síntomas o consecuencias de la enfermedad o que, en el mejor de los casos, logre curarla. En mi laboratorio del Centro Nacional de Biotecnología investigamos sobre albinismo, una condición genética poco frecuente que afecta a unos tres mil españoles, caracterizada por una discapacidad visual severa, presente en todos los casos, y por alteraciones pigmentarias con una manifestación variable. Conocemos una veintena de genes cuyas mutaciones causan otros tantos tipos de albinismo. Detectar en cada caso cuál es el gen afectado y cuáles son las mutaciones responsables se ha convertido en un reto al que nos dedicamos desde hace más de diez años, desde el CIBERER, y en colaboración con grandes genetistas del país como Ángel Carracedo (USC, Santiago de Compostela) y Carmen Ayuso (FJD, Madrid).
Detectar el gen causante de albinismo nos puede llevar unas semanas o muchos años, en función de la suerte que tengamos en detectar las mutaciones con las diferentes estrategias metodológicas que aplicamos. Pero, una vez lo encontramos, una vez detectamos al gen responsable del albinismo de una persona es muy gratificante ver la emoción que suscita en las familias. No hemos conseguido nada. No hemos curado la enfermedad rara. Pero ahora sabemos a qué nos enfrentamos. El desconocimiento produce desazón, angustia, desasosiego. Saber qué nos pasa nos reconforta. Esto puede ser difícil de entender para quien no haya vivido estas situaciones, pero en toda familia en la que se instala una enfermedad rara su lucha inicial se centra en conseguir un diagnóstico, lograr saber por qué aquel hijo o hija ha nacido con esa enfermedad poco frecuente, inesperada. Y la alegría contenida que se libera cuando remitimos ese informe genético y llega a manos de la familia es una recompensa enorme, que nos aporta la motivación necesaria para continuar investigando. También resulta alentador descubrir las iniciativas de padres emprendedores que desarrollan estrategias para mejorar el proceso de diagnóstico genético o los proyectos europeos que se crean con objetivos similares.
¿Existen terapias para tratar las enfermedades raras? El consorcio internacional de investigación en enfermedades raras (IRDiRC) tiene entre sus objetivos para la década 2017-2027 lograr diagnosticar genéticamente todos los pacientes con enfermedades raras en menos de un año y desarrollar por lo menos 1.000 terapias para el tratamiento de enfermedades raras para las cuales hoy en día no tenemos cura posible. Los tratamientos pueden involucrar la administración de medicamentos, de pequeñas moléculas con actividad terapéutica. O el reposicionamiento de fármacos originalmente aprobados para tratar una enfermedad pero que, con el tiempo, resultan ser también efectivos para tratar otras enfermedades. O mediante terapia génica clásica, añadiendo las funciones génicas que faltan. Éxitos recientes en nuestro país, como la terapia génica para tratar a los niños con anemia de Fanconi, desarrollada por el grupo de Juan Bueren del CIEMAT-IIS FJD-CIBERER, permiten albergar esperanza de que los beneficios podrán ampliarse a otras patologías usando estrategias similares.

El futuro se llama edición genética. La posibilidad de cambiar a voluntad las letras incorrectas de nuestros genes, gracias a las herramientas CRISPR, y el poder hacerlo con seguridad y eficacia, será uno de los apasionantes retos científicos y tecnológicos que afrontaremos durante los próximos años. Y para ello, antes de llegar a la clínica, deberemos seguir explorando nuevas ideas terapéuticas usando modelos celulares o animales de estas enfermedades raras, que nos permitan avanzar en nuestro entendimiento de por qué aparece la enfermedad y qué podríamos hacer para evitarlo. Ya estamos siendo testigos de los primeros éxitos logrados para tratar pacientes de enfermedades graves de la sangre, como la anemia falciforme o la beta-talasemia. O para tratar pacientes de cáncer refractarios, mielomas o sarcomas recalcitrantes que ahora pueden ser tratados con estrategias innovadoras de inmunoterapia con la participación de la edición genética.
Me gustaría terminar esta tribuna de opinión recordando el cuarto principio de bioética: el principio de justicia. Nada de todo lo anterior tendrá sentido si no ponemos las terapias que seamos capaces de imaginar y validar al alcance de todas las personas que lo necesiten, sin distinción de ningún tipo. Por ello deberemos emplearnos a fondo para lograr compaginar el legítimo derecho que tienen las empresas farmacéuticas para recuperar las inversiones realizadas, con el derecho que tienen los pacientes de ser tratados con los tratamientos más efectivos, y el deber de las autoridades para que todo ello ocurra. Habrá que explorar otros escenarios, como los basados en riesgos compartidos o en el valor terapéutico de los medicamentos.
Este artículo lo publiqué inicialmente en la Agencia SINC el 28 de febrero de 2020.
Mi hijo de 16 años padece hipopotasemia familiar severa de primer grado desde los 15 años y hoy está con parálisis en algunas partes del cuerpo. Hoy tengo que ir a trabajar y no me va a ser posible ya que vivimos solos y tampoco puedo pedir ayuda porque se asustan. Que derecho como trabajadora tengo si falto hoy…..estoy desesperada porque la ley de dependencia aún está a trámite sin cursar por circunstancias del covid y yo trabajo en un hospital en otra ciudad. No se como afrontar todo esto pues ya estuvo en UCI y mi miedo es que me sancionen en el trabajo pues estoy cubriendo una IT. Por otro lado me gustaría saber de familias que tengan este problema similar para poder contrastar. Un saludo
Gracias por compartir tu caso Fini. Te recomiendo que te pongas en contacto con FEDER (https://enfermedades-raras.org/) para intentar encontrar otras familias con casos similares que puedan haberse organizado ya constituyendo una asociación, que sería la que podría prestarte la ayuda que necesitas. Adicionalmente las administraciones (especialmente las CCAA y los ayuntamientos) cuentan con servicios sociales que se pueden solicitar para casos como el tuyo. Te deseo que encuentres una solución cuanto antes. Cordiales saludos. Lluís
Tengo una hija de 17 años de edad y tiene una discapacidad por una trisomia 9 y monosomia 7 quisiera saber si tienen conocimiento de algún caso similar gracias
Puede preguntar en CIBERER (https://www.ciberer.es/) o en FEDER (https://enfermedades-raras.org/), ambas instituciones tienen servicios para buscar otros pacientes con patologías similares.
Pueden ayudarme con información sobre el diagnóstico en una niña con diagnóstico asociación murcS