
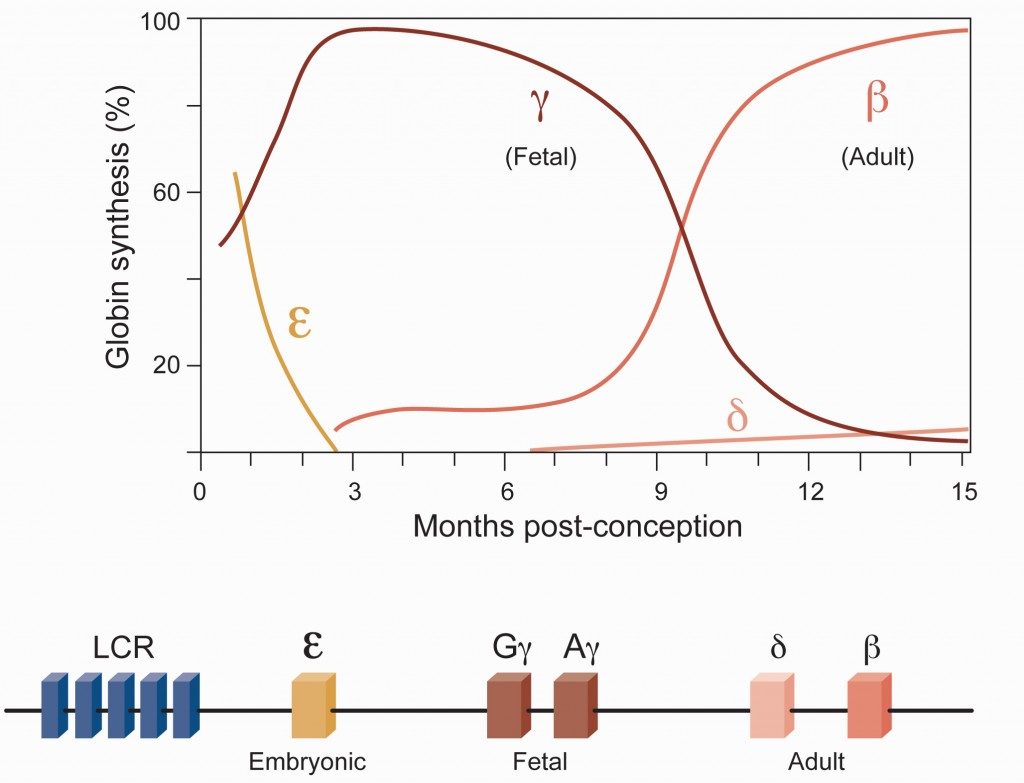
La hemoglobina es la proteína principal de los glóbulos rojos de la sangre y es la responsable, entre otras funciones y gases que puede transportar, de llevar el oxígeno (O2) que necesitan todas las células del cuerpo para seguir funcionando . La hemoglobina en adultos está formada por dos subunidades, alfa y beta, dos de cada, dos cadenas de alfa-globina y dos cadenas de beta-globina. Cada subunidad de globina está codificada por genes distintos. En relación al locus de la beta-globina no existe un solo gen sino varios, agrupados en una misma región del genoma, que se van turnando, activándose y desactivándose durante el desarrollo embrionario, fetal y en adultos. Como aparece en la primera imagen de este artículo, del laboratorio de Daniel Bauer, la globina embrional se usa durante los primeros tres meses de gestación, y es reemplazada entonces por la globina fetal que finalmente es substituida por la beta-globina adulta tras el nacimiento.
Existen enfermedades graves de la sangre (como la anemia falciforme o la beta-talasemia) causadas por mutaciones en la secuencia del gen de la beta-globina adulta (anemia falciforme, con 300.000 nuevos casos detectados cada año en todo el mundo, mayoritariamente en África y en India) o por mutaciones a lo largo de todo el gen de la beta-globina, incluidas las zonas reguladoras [como la zona LCR de la primera imagen], no codificantes, que determinan un déficit en la expresión de beta-globina (beta-talasemia, con 50.000 nuevos casos diagnosticados anualmente, relativamente frecuente en países del Mediterráneo y en África), y que interfieren con la capacidad de transportar oxígeno de la hemoglobina y provocan un cuadro de síntomas clínicos graves que requiere tratamiento médico, fundamentalmente transfusiones de sangre de donantes sanos.
Una alternativa terapéutica a las transfusiones sería reactivar la globina fetal, inactiva en adultos, para que supliera la variante mutante de beta-globina adulta, que es la que porta la mutación. Para ello se han explorado diversas estrategias y tratamientos, pero la que diseñó y describió el laboratorio de Daniel Bauer, en este artículo publicado en la revista Nature Medicine en mayo de este año, es la que parece haber triunfado. Se trata primero de todo, de saber por qué no funciona el gen de la globina fetal en adultos. No funciona porque está reprimido, frenado, por un represor. Una proteína represora llamada BCL11A, codificada por un gen del mismo nombre, que mantiene apagado el gen de la globina fetal en la vida adulta. Siendo así, si reprimimos al represor potenciaremos lo reprimido, reactivando así el gen de la globina fetal.
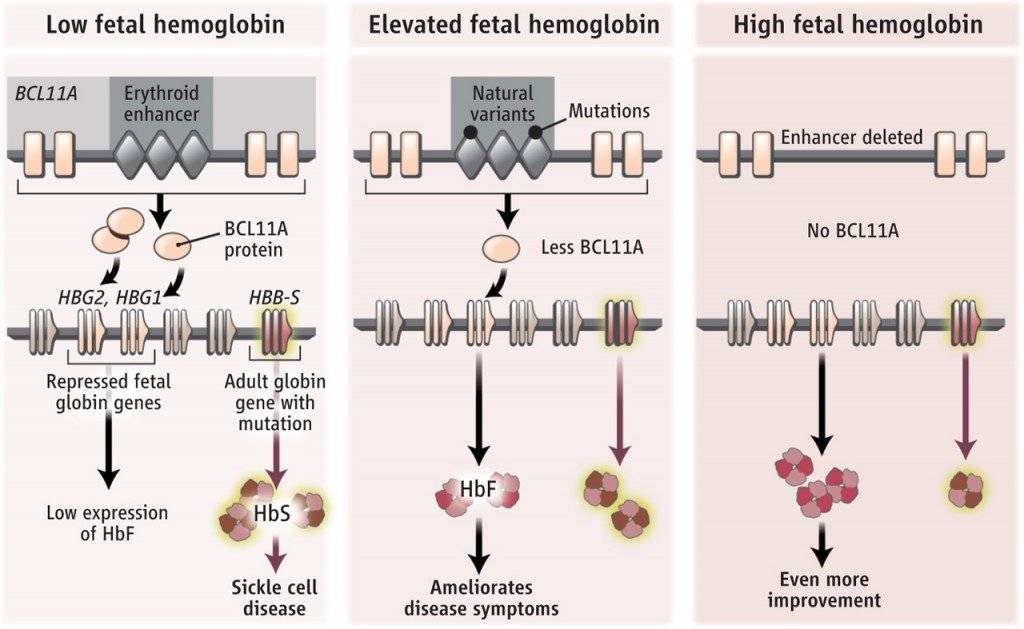
El equipo de Daniel Bauer conocía que hay algunas personas que son portadoras de mutaciones en una región reguladora (un potenciador de la expresión, un enhancer, en inglés) del gen BCL11A que determinan una menor expresión de este (una menor producción del represor BCL11A), y, a la postre, un incremento de la globina fetal, que deja de estar reprimida como debiera en adultos. Y se les ocurrió usar la tecnología CRISPR-Cas9 para eliminar esta región reguladora, limitando así de forma importante la expresión del gen BCL11A y reprimiendo su actividad represora, con lo que, de nuevo, conseguirían reactivar la expresión de los genes fetales de globina. Es importante reseñar que no se puede eliminar el gen BCL11A sin más, al ser necesario para el desarrollo de otras células de la sangre. Lo que hace esta aproximación novedosa es reducir de forma significativa su expresión, reducir/reprimir al represor. Para que tenga efecto sobre los genes de las globinas fetales pero que todavía pueda seguir actuando donde se le necesita en otros genes.
Se realiza la eliminación, la deleción de esa zona reguladora en las células madre de la sangre (células troncales hematopoyéticas, HSC) extraídas del paciente afectado (por anemia falciforme o beta-talasemia), en el laboratorio, y, tras verificar la deleción correcta, se reinfunden estas mismas células editadas en el mismo paciente, para que generen ahora glóbulos rojos con hemoglobina que tendrá una cadena alfa y otra cadena fetal de globinas, que suplirá a la cadena beta-globina mutada. Una estrategia muy imaginativa e innovadora: reprimir al represor.
En colaboración con dos empresas del sector: CRISPR Therapeutics y VERTEX, que aportaron los reactivos CRISPR-Cas9 necesarios para eliminar esos elementos reguladores de ADN, y con la colaboración de varios hospitales, se registraron dos ensayos clínicos, para evaluar seguridad y eficacia para tratar a pacientes con anemia falciforme y con beta-talasemia con esta estrategia terapéutica, denominada técnicamente: CTX001, una terapia génica ex-vivo basada en las herramientas CRISPR-Cas9 de edición genética.
Los resultados que acaban de conocerse de los primeros dos pacientes tratados, uno con anemia falciforme y el otro con beta-talasemia, son muy positivos. En ambos casos han desaparecido los síntomas clínicos de las enfermedades de la sangre, y ya no necesitan más transfusiones, lo cual es todo un logro científico y médico lo que se ha conseguido con esta terapia génica ex-vivo basada en CRISPR, uno de los primeros éxitos palpables de la aplicación de estas herramientas en el ámbito hospitalario, para tratar pacientes de enfermedades congénitas graves.
Habrá que esperar a tener datos a más largo plazo, de estos y de otros pacientes, para confirmar la universalidad, seguridad y eficacia de este nuevo tratamiento basado en CRISPR. Pero sin duda es una magnífica noticia, para todo el sector, desde los investigadores, a los médicos y a los biotecnólogos de las empresas farmacéuticas encargadas de fabricar los reactivos CRISPR necesarios. Enhorabuena a todos!.
Hace tiempo le escuché en una entrevista diciendo que las terapias CRISPR curativas tardarían un tiempo en llegar, pero que si veríamos relativamente pronto el CRISPR utilizado como detector de enfermedades. Con estos nuevos avances, ¿Piensa que ya hemos entrado en la fase curativa?
Hola Álvaro, sigo pensando que la aplicación universal y sistemática de estrategias terapéuticas CRISPR en la clínica todavía tardará algún tiempo, pero está claro que este ejemplo nos invita a pensar que las terapias génicas ex-vivo puede que tengan una transición más corta y exitosa que el resto de terapias in-vivo.
cordiales saludos.
Estoy investigando que otro tratamiento parenteral puede estimular la médula ósea
Para leucemia mkeloide
Entiendo que lo más significativo de este trabajo es encontrar una diana para el sistema CRISPR Cas-9 que sea eficaz contra la enfermedad y no produzca otros daños, pero mi pregunta es: antes de aplicarla a pacientes es preciso experimentar con animales para comprobar su eficacia y seguridad?