

Esta semana pasada la revista Science finalmente ha publicado un estudio que supuestamente concluía que completaba la secuencia del genoma humano. Vayamos por partes. En realidad ese estudio lo conocimos hace casi un año, cuando sus autores lo depositaron en el servidor de preprints bioRxiv, compartiendo sus hallazgos y poniéndolos generosamente a disposición de la comunidad científica. Esta es la tónica actual, que empodera a los investigadores, a despecho de las revistas, que ven como dejan de poder controlar las noticias de avances en el conocimiento, decisión que ahora toman los investigadores al compartir su trabajo, mientras inician la árdua y frecuentemente imprevisible trayectoria por diversas revistas y editoriales hasta dar con la que acepta publicar el artículo, habitualmente con diversas modificaciones solicitadas por los revisores del trabajo. Algunas de estas sugerencias naturalmente mejoran la calidad del trabajo pero otras, sencillamente, reorientan y desenfocan el mensaje original de los autores, que acaban aceptando algunos cambios para garantizar la publicación de sus resultados. Por eso es tan importante poder disponer de los trabajos originales, tal y como se diseñaron, ejecutaron e interpretaron por los autores.
Tras la publicación (en bioRxiv, en mayo de 2021) de esa extensión del conocimiento de nuestro genoma fuimos varios quienes comentamos la relevancia del estudio y su impacto. Yo mismo escribí una tribuna para la agencia SINC, que también recogí en este blog, y en este programa de A hombros de gigantes, en RNE, con Manuel Seara. Ahora, finalmente con la publicación del estudio en la revista Science, dentro de un número especial con diversos trabajos complementarios, ha renacido el interés por el trabajo, que ha vuelto a los titulares de los medios. Y, naturalmente, he vuelto a colaborar con los medios comentando nuevamente ese estudio y sus derivadas genéticas con Sergio Ferrer, Mario Viciosa, Marcos Domínguez y Luis Alfonso Gámez, entre otros. Pero dado que veo que sigue habiendo un cierto debate sobre si hemos completado o no el genoma humano y sobre lo que ha conseguido este consorcio de investigadores he creido oportuno publicar esta nueva entrada cuyo mensaje está incluido en el título: ahora conocemos mejor nuestro genoma, aunque no todavía del todo.
Las palabras de aquellos que tenían dudas y manifestaron sus críticas ante el proyecto genoma, dudando sobre si merecía la pena secuenciarlo o no, cuando se lanzó la iniciativa a principios de los años 90 del siglo pasado, se las llevó el viento. Está claro que uno de los hitos científicos más importantes de la biología, y de la ciencia en general, es haber podido conocer y descifrar el genoma de nuestra propia especie. Sin embargo, este ha sido un reto complejo, que ha ido completándose progresivamente, de forma incremental, y que, sorprendentemente todavía no ha terminado. Para el gran público, para los medios de comunicación, quedan las publicaciones de febrero de 2001, como las que recordamos para referirnos a la publicación del genoma humano. Sin embargo, esas primeras publicaciones contenían solamente un primer borrador, con muchas zonas todavía indeterminadas (verdaderos agujeros en la secuencia) que tuvieron que ir siendo rellenados con esfuerzo. Así llegamos a febrero de 2003, una fecha habitualmente no incluida en las efemérides genómicas, pero en la que realmente se dio un salto significativo en la calidad del genoma humano secuenciado, completándose muchos de los agujeros que todavía permanecían sin rellenar. Tras esa fecha, el genoma humano ha seguido revisándose y ampliándose, corrigiendo errores de ensamblaje y ampliando las zonas que todavía permanecían indeterminadas, hasta llegar a la versión de 2013, denominada técnicamente GRCh38.p13 que es la que hemos seguido usando hasta ahora, por ejemplo para identificar las mutaciones que presentan los pacientes de enfermedades raras congénitas, comparando el genoma de estos con el de referencia. Y estos investigadores ahora lo que han hecho es mejorarla, ampliarla, aumentar nuestro conocimiento del genoma de forma substancial, aunque sin completarlo del todo todavía.
Los 20.000 genes que tenemos apenas ocupan un 2% de nuestro genoma. Mientras que el 98% restante es lo que llamamos el genoma no codificante, o intergénico. Esta parte mayoritaria del genoma está formada por diversas secuencias repetidas, transposones y retrotransposones de diversas familias y también contiene los elementos reguladores de la expresión génica, los interruptores que les indican a los genes en qué célula deben funcionar y cuál no, y cuándo durante el desarrollo deben encenderse y apagarse. Y es ahí donde tenemos la mayor parte de los agujeros e indeterminaciones. Imaginad por ejemplo una zona que tuviera la secuencia TA repetida muchas veces (TATATATATATATATATA….). Imaginad que hubiera 500 repeticiones. Si estamos leyendo fragmentos de 100-200 letras, ¿cómo vamos a saber que aquí hay 500 repeticiones de TA y no 200, o 300, u 800?. Solo lo sabremos si somos capaces de leer una misma molécula de ADN, que empiece antes de las repeticiones y termine después de ellas, para leer toda esta zona repetitiva de cabo a rabo.
¿Qué importancia tiene no disponer de un genoma humano completo? Realmente es preocupante no tener toda la información genética de una especie, en este caso de la nuestra, de los seres humanos. Más allá de que nos perdemos información que podría contener genes de los cuales desconocemos su existencia las implicaciones prácticas de esas lagunas en el conocimiento genómico se manifestan a la hora de diagnosticar genéticamente pacientes que clínicamente los asociamos a una determinada patología pero que, cuando revisamos su genoma, no somos capaces de encontrar la causa molecular, la mutación causante de esa enfermedad. Esto es así para alrededor de un 30% de pacientes de cualquiera de las miles de enfermedades raras de base genética. Para ellos seguimos siendo incapaces de determinar por qué están enfermos. Pero claro, si desconocemos una parte del genoma humano no podemos descartar que la posible mutación que buscamos pueda estar oculta en esas zonas que siguen siendo oscuras para los análisis, en las que no podemos investigar. Simplemente porque las desconocemos. Por ello, al rellenar muchas de estas zonas indeterminadas, además de completar nuestro genoma, estamos dándoles la oportunidad a todos esos pacientes no diagnosticados de que pueda encontrarse las mutaciones causantes de sus enfermedades.

Los métodos de secuenciación masiva actuales progresan obteniendo la información de fragmentos pequeños del genoma, de 100 a 200 letras, aproximadamente, millones de ellos, que finalmente un programa bioinformático tiene que ensamblar y colocar en su posición en el genoma. Cada uno donde le corresponde. Viene a ser como resolver un puzle de muchas piezas, pongamos 10.000 piezas. Cuando nos disponemos a hacer un puzle de estas características nuestro principal temor es que se hayan olvidado alguna pieza que nos impida completar la imagen completa que se oculta tras el puzle. Pues con la secuenciación masiva del genoma pasa justamente lo contrario. No nos faltan sino que nos sobran piezas. Hay miles de fragmentos que no sabemos donde los tenemos que colocar, pues una vez completado todo el proceso de ensamblaje (una vez rellenadas todas las piezas del puzle) nos siguen quedando piezas que no sabemos qué hacer con ellas. En esta entrada anterior sobre nuestro genoma explicaba los casi 200 millones de letras que habitualmente tenemos que guardar en un cajón, pues nuestro genoma de referencia sigue teniendo agujeros, sigue sin estar completo. Al ampliar el genoma añadiendo casi 200 millones de letras más este estudio habría ampliado la foto de nuestro puzle, generando agujeros nuevos (resolviendo secuencias) en los que ahora podemos ubicar muchos (pero no todos) de los fragmentos que antes iban a parar al cajón. Y esto es genial, muy importante. Ahora sabemos mucho más de nuestro genoma, aunque sigamos sin tener el conocimiento exhaustivo, completo y definitivo del mismo.
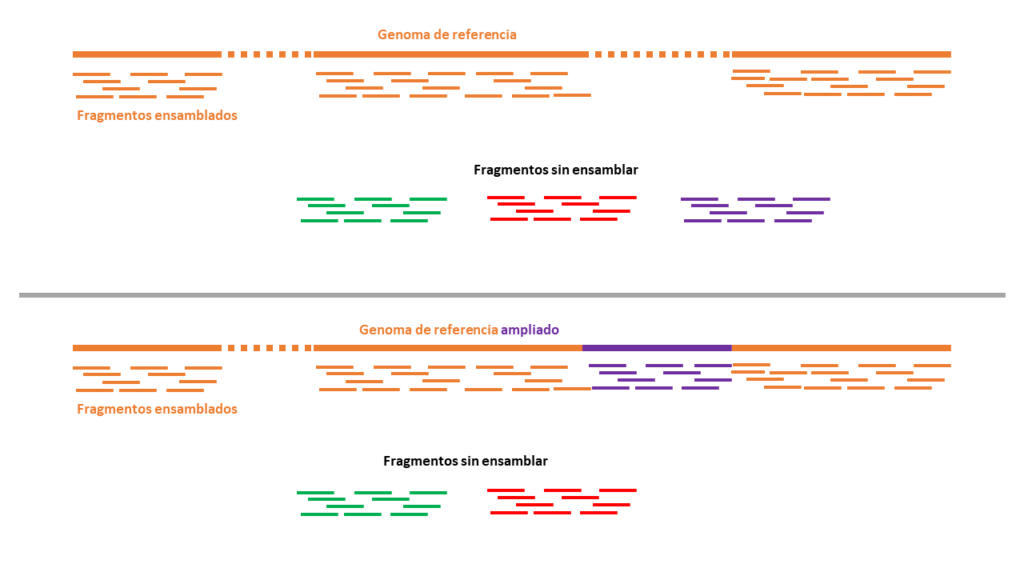
En concreto, este estudio ha usado diversos trucos experimentales para completar estos agujeros del genoma, que ya explicaba en mi entrada anterior. Para empezar ha usado células (CHM13) derivadas de patologías placentarias que se caracterizan por tener solamente los cromosomas de uno de los progenitores, frecuentemente el femenino. Tienen 23 cromosomas, pero solo una copia de cada par o, si tienen dos copias, ambas son idénticas, estando todo el genoma en homocigosis. Es lo que se llama células funcionalmente haploides. Esto hace que su secuenciación sea menos problemática, al no tener que enfrentarse a descifrar las secuencias similares, pero no idénticas, que tenemos todos nosotros en nuestras células, con una copia de cada gen recibida de nuestro padre y la otra de nuestra padre, que pueden contener diferencias. Claro, al ser de origen femenino carecen del cromosoma Y. Por lo tanto, toda la información que nos faltaba rellenar del cromosoma Y nos sigue faltando. Aquí tenéis la explicación de por qué todavía no hemos completado el genoma humano. Además del resto de agujeros todavía no resueltos que siguen quedando en el genoma, y de regiones intrínsecamente difíciles de descifrar, por su alto grado de repeticiones, que siguen sin ser establecidas inequívocamente, como reconocen los propios autores. Otro de los trucos que han usado ha sido aplicar nuevos métodos de secuenciación (PacBio, NanoPore). Por ejemplo NanoPore es un sistema que permite secuenciar moléculas largas de ADN que se hacen pasar a través de una minúscula apertura, un nanoporo, como si enebráramos una aguja con un hilo muy largo y lo hiciéramos pasar por el agujero sin romperlo. Estas técnicas permiten superar las limitaciones de la secuenciación basada en fragmentos pequeños, pero tienen la limitación de que acumulan muchos más errores que las tecnologías anteriores, lo cual también hay que tener en cuenta.
Idealmente lo que nos gustaría es poder pasar a través de una máquina de secuenciación cada uno de los 24 cromosomas (del 1 al 22, el X y el Y) que caracterizan nuestra especie, leyendo todas y cada una de las letras, con precisión y robustez. Y entonces sí que podríamos decir que hemos completado el genoma humano. Pero todavía no disponemos de la tecnología necesaria para poder secuenciar cromosomas completos y tenemos que conformarnos con las limitaciones de las tecnologías actuales.
Los resultados principales de esta nueva ampliación del genoma humano son impactantes, y han mejorado mucho nuestro conocimiento del genoma. Han añadido un 8% más de letras a nuestro genoma, casi 200 millones de letras más, en las que los ordernadores les dicen que podrían haber unos 2.000 genes, 99 de los cuales parecen codificar proteínas. Esto es relevante. Son casi 100 genes de los que desconocíamos su existencia, y que habrá que añadir a los que conocemos. Y cuyas mutaciones también pueden ser la causa de algunas enfermedades, ayudando a completar el diagnóstico genético de muchos pacientes actualmente no diagnosticados.
Finalmente, otra de las limitaciones importantes de este estudio es que no aporta información sobre la diversidad de genomas humanos que existen. A pesar de que habitualmente usamos un solo genoma de referencia lo cierto es que acumulamos diferentes variantes genéticas que son características de diferentes poblaciones, y es importante conocerlas para interpretar correctamente los resultados de la secuenciación del genoma de una persona. Actualmente ya existen genomas de referencia para diferentes poblaciones (que se ensamblan usando el genoma actual de referencia, el de 2013), pero habrá que ampliarlos usando estas mismas tecnologías, para que los beneficios de esta ampliación no solo lleguen a un genoma (al que tienen las células usadas en este estudio) sino a múltiples genomas, de diferentes grupos poblacionales, lo cual es esencial para dilucidar si una variante es patogénica (causante de enfermedad) o es simplemente una variante polimórfica característica de esa población.
Ahora conocemos mejor nuestro genoma, aunque no todavía del todo.
Super excelente artículo, leído por un médico no genetista, ideal para información del público.
Simplemente soy una curiosa anónima que desde que en bachillerato nos mostraron la célula se apasionó con la biología. Me hubiera gustado estudiar genética pero nadie fue capaz de guiarme.
Ahora, por desgracia, soy paciente oncológico desde hace ventisiete años y sigo todas las noticias que me parecen relevantes. Y esta lo es, y mucho.
Muchas gracias por su tiempo y atención.
En relación a la formación de la mola hidatiforme completa, no me queda claro su origen. Por lo que tengo entendido la mola hidatiforme completa se forma a partir de un óvulo que ha perdido su genoma y ha sido fecundado por un espermatozoide que puede ser 23X o 23Y. En el caso de que haya sido fecundado por un espermatozoide 23X, el pseudoembrión carecerá de cromosoma Y, pero, precisamente lo que conserva es el ADN parental y no el maternal.
El hecho de proponer como nuevo genoma de referencia, un genoma procedente de una línea celular continua (trofoblastos CHM13), con todas las mutaciones que haya podido ir acumulando a lo largo de los diferentes pases in vitro, ¿no será contraproducente para tomarlo como referencia de normalidad?
Siempre, gracias por su artículo.
Hola Javier, son muchas las circunstancias biológicas que pueden dar lugar a las líneas celulares CHM.
En concreto para la línea celular CHM13, que es la que han usado en este proyecto T2T, puedes encontrar más información en estos sitios web:
https://web.expasy.org/cellosaurus/CVCL_VU12
https://sites.google.com/ucsc.edu/t2tworkinggroup/chm13-cell-line
El genoma actual humano también es producto de diversos orígenes, también de diversas líneas celulares.
La secuencia genomica de 2003 se estableció a partir del ADN obtenido de leucocitos de unos cuantos donantes.
¿Es la secuencia de 2013 la que se ha obtenido a partir de alguna línea celular continua?
Como comento en el artículo la ampliación del genoma se ha hecho a partir de la secuencia de la línea celular CHM13
Gracias por el blog, me es muy apasionante enterarme de éstos avances… Soy estudiante de biología molecular… Me gustan sus videos, Saludos desde México